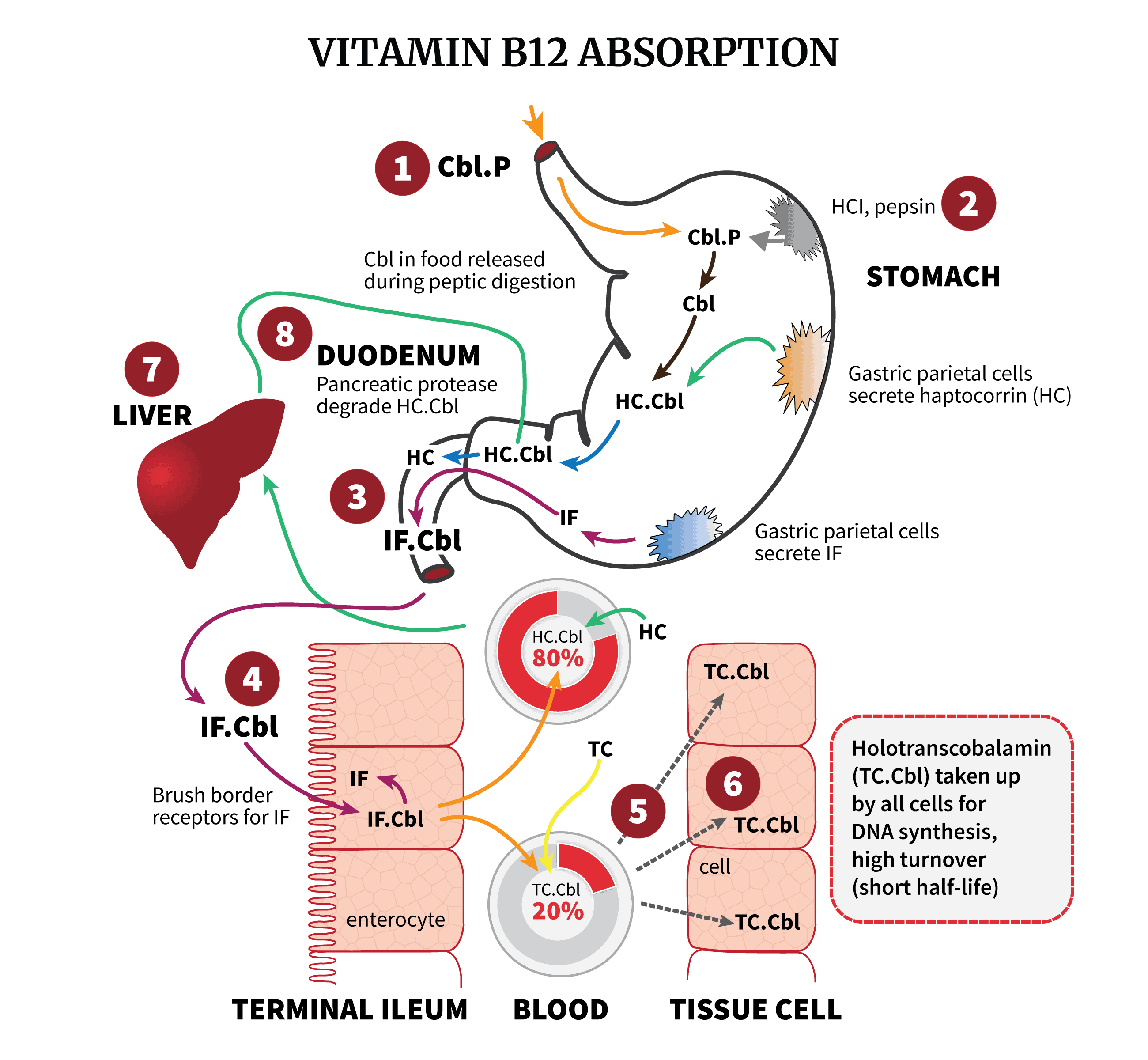
Transcobalamins are carrier proteins which bind to cobalamin (B12).
The first type is transcobalamin-1 (referred to as haptocorrin, or HC), which the salivary glands create. It helps protect the B12 from stomach acid degradation. Once the HC-B12 complex has traveled to the duodenum, pancreatic proteases release the B12, which then binds to intrinsic factor (IF) for absorption by ileal enterocytes. After it has crossed into the ileum, the IF-B12 complex degrades, and transcobalamin-2 (referred to simply as transcobalamin, or TC) binds to the B12 and transports it to cells and tissues.
Perhaps we can better explain it visually, through an illustration taken from our article about cobalamin absorption (which we highly recommend to read):
Obviously, we’re here to discuss the deficiency of TC, which – although rare – could lead to B12 deficiency. This TC deficiency doesn’t necessarily mean transcobalamin is totally absent. It could also mean transcobalamin is not functioning correctly.
Recent epidemiologic studies suggest that not only the amount but also the regular functioning of TC-II is important for normal development of the nervous system. Loss of TC-II affinity for vitamin B12 in mothers was associated with a three to fivefold increased frequency of neural tube defects in fetuses.
Three mechanisms of TC-II deficiency have been reported: common type is characterized by the absence of both B12 binding components and immunoreactive TC-II; in the second type there is a lack of B12 binding components but normal immunoreactivity; and in the third type TC-II binds B12 but is non-functional.
Normally, this is an autosomal recessive inheritance discovered in infancy (autosomal recessive means, if baby boy has such a gene – both parents carry and had inherited it to him), and in which B12 injection treatment into adulthood is necessary. This recessive, complete TC-II deficiency is characterized by severe megaloblastic anemia, intestinal abnormalities and immunodeficiency, and neurological dysfunction including mental retardation, ataxia, pyramidal deficit and seizures. It is a serious condition:
TC deficiency is a rare autosomal recessive disorder usually presenting in early infancy with failure to thrive, weakness, diarrhea, pallor, anemia, and pancytopenia or agamma-globulinemia. It can sometimes resemble neonatal leukemia or severe combined immunodeficiency disease. Diagnosis of TC deficiency is suspected based on megaloblastic anemia, elevation of total plasma homocysteine, and blood or urine methylmalonic acid. It is confirmed by studying the synthesis of TC in cultured fibroblasts, or by molecular analysis of the TCN2 gene.
Transcobalamin (TC) deficiency is a rare autosomal recessive inborn error of cobalamin transport which clinically manifests in early infancy. We describe a child with TC deficiency who presented with classical clinical and lab stigmata of inborn error of vitamin B12 metabolism except normal serum B12 levels. He was started on empirical parenteral cobalamin supplements at 2 months of age; however, the definitive diagnosis could only be established at 6 years of age when a genetic evaluation revealed homozygous nonsense variation in exon 8 of the TCN2 gene.
Transcobalamin II deficiency is a rare autosomal recessive disorder causing intracellular cobalamin depletion, which in turn causes megaloblastic bone marrow failure, accumulation of homocysteine and methylmalonic acid with clinical findings of failure to thrive, diarrhea, vomiting, pancytopenia, megaloblastic anemia, and neurological findings. Homozygous or compound heterozygous mutations in the transcobalamin II gene on chromosome 22q12.2 that contains 9 coding exons are known to cause transcobalamin II deficiency, including deletions, nonsense mutations, and a mutation resulting in activation of a cryptic intronic splice site. Herein, we describe the clinical findings at presentation and outcome of 4 patients with genetically confirmed novel transcobalamin II gene mutations, of whom 3 had large deletions of 1 kb and 1 had a homozygous Q36X mutation.
In infants with pancytopenia, growth retardation, gastrointestinal manifestations, and immunodeficiency, the inborn error of cobalamin metabolism should be kept in mind. Early diagnosis and treatment are crucial for better clinical outcomes. What is new? In literature, to date, less than 50 cases with TC deficiency were identified. In this report, we presented twins with TCN2 gene mutation. Both patients emphasized that early and aggressive treatment is crucial for achieving optimal outcomes. In this report, we identified a novel variation in TCN2 gene.
A male Caucasian infant presented at 6 weeks of age with failure to thrive, diarrhoea, macrocytic anaemia, and decreased IgG. He had normal serum B12 and folate levels. Serum cobalamin binding capacity showed no detectable transcobalamin II. Both parents showed levels consistent with a heterozygous state. The literature is extensively reviewed, and the importance of early diagnosis to prevent neurological dysfunction is stressed.
Even when symptoms are discovered later in life, the situation could still be traced back to an undiagnosed, autosomal recessive inborn error of B12 metabolism:
Transcobalamin deficiency is a rare autosomal recessive inborn error of cobalamin transport (prevalence: < 1/1000000) which clinically manifests in early infancy. We describe the case of a 31 years old woman who at the age of 30 days presented with the classical clinical and laboratory signs of an inborn error of vitamin B12 metabolism. Family history revealed a sister who died at the age of 3 months with a similar clinical syndrome and with pancytopenia. She was started on empirical intramuscular (IM) cobalamin supplements (injections of hydroxocobalamin 1 mg/day for 1 week and then 1 mg twice a week) and several transfusions of washed and concentrated red blood cells. With these treatments a clear improvement in symptoms was observed, with the disappearance of vomiting, diarrhea and normalization of the full blood count. At 8 years of age injections were stopped for about two and a half months causing the appearance of pancytopenia. IM hydroxocobalamin was then restarted sine die. The definitive diagnosis could only be established at 29 years of age when a genetic evaluation revealed the homozygous c.1115_1116delCA mutation of TCN2 gene.
But is that always the case? Rather than being an autosomal recessive disorder whose symptoms are present from infancy, could TC-II deficiency also be an autosomal dominant disorder (passed on by just one parent), with milder presence?
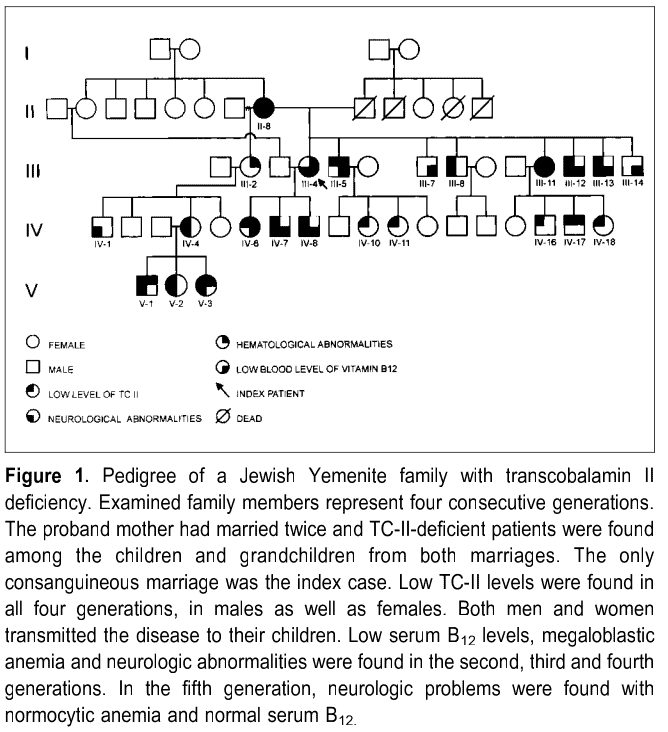
In 2004, Dr. Valery Teplitsky discovered a Jewish family of Yemenite origin with what seems like an autosomal dominant hereditary partial transcobalamin II deficiency disorder. He found low apo TC-II (free TC-II unbound to B12) and low total unsaturated B12 binding capacity in all tested individuals but one, and low holo TC-II (TC-II bound by vitamin B12, also known as active B12) in all family members.
We present here a Jewish family of Yemenite origin with neurologic and hematologic manifestations associated with partial TC-II deficiency transmitted in a dominant pattern. This family workup suggests that the heterozygous state of TC-II deficiency may also be of clinical importance.
In contrast to the autosomal-recessive mode of inheritance in previously described families with TC-II deficiency, in our family the transmission of the disease seems to be autosomal-dominant, although the x-linked dominant type of inheritance could not be excluded. The possibility of a TC regulatory gene abnormality rather than a TC-II genetic defect has not yet been investigated, although such a mutation in this putative gene might explain the clinical picture in some historical cases.
In our family, partial TC-II deficiency was the main biochemical abnormality and the most probable cause of the second type of neurologic complications ± the complex of symptoms resembling minimal brain dysfunction.
The family above is unique, because in contrast to the known autosomal-recessive complete TC-II deficiency, they all had TC-II present, albeit in a low amount. Still, their partial transcobalamin deficiency was associated with clinically significant neurological and hematological complications. That milder degree of TC deficiency could explain the lack of symptoms or the late appearance in some of the members. Recessive vs. dominant transmission could be the difference between partial and complete TC-II deficiency.
How common is this adult onset partial transcobalamin II deficiency? Well, the Jewish Yemenite family is probably not alone. For example, one of our readers reports having had low transcobalamin progressing to zero transcobalamin. She writes:
My siblings are all approaching middle age. I was the first to drop from Low Transcobalamin to Zero Transcobalamin that landed me in the Emergency Room three times for suspected stroke and suspected multiple sclerosis in the month of January 2023. The drop from Low Transcobalamin to Zero Transcobalamin happened to me within the short time of just 1 month, at age 44. I really feel I need to figure this ‘Transcobalamin Deficiency’ out so that I can convince my siblings to do the blood test and pre-emptively inject Methyl-B12 into themselves before the drop to Zero Transcobalamin occurs in their bodies, so that they don’t die and so that they don’t have irreversible brain damage.
We know she doesn’t have the classic autosomal recessive form of TC deficiency, because her tests from Invitae came back negative for the known genes. As a side note, she doesn’t have genetic intrinsic factor deficiency either, and her MTHFR gene tests with Aventus show “MTHFR (A1298C) Heterozygous” and “MTHFR (C677T) Normal”.
Furthermore, our reader’s symptoms, blood profile, and family history all match exactly the family from Dr. Teplitsky’s study. She even suspects it may have something to do with being consanguineous (descendants of an inter-familial union between second-cousins or closer), since the index case in Dr. Teplitsky’s study was married consanguineous, and our reader has first-cousin marriages in both her father and mother side.
These are speculations. It could also be that some undiscovered genes are affecting the gut-brain-immune axis in patients with unique profiles of TC-II deficiency. We don’t know. But this condition isn’t new; TC deficiency has been referenced long ago, such as in the obscure book Symposium On Enzyme Defects And Immune Dysfunction from 1978.
What to Do?
In conclusion, even though TC-II deficiency is rare, it should be suspected in individuals with megaloblastic anemia or neurological and cognitive deficits. Early detection in infancy or childhood is important, allowing B12 deficiency treatment early on, to support normal neurological development and prevent irreversible disability.
Whether you have true, autoimmune pernicious anemia or transcobalamin II deficiency of any of the types described, the treatment is the same: daily B12 injections. For the vast majority of people, we highly recommend the methyl B12 form. If your doctor refuses to prescribe them on a daily basis, you could get a month-worth of shots from us for $129 or less, instead of paying $50-250 a shot in private clinics.